


From directives to regulations, legislators have comprehensively overhauled compliance requirements for medical device manufacturing companies selling to the European market.
In this article, we’ll review the regulations and directives for medical devices and explain how manufacturers can stay compliant.
The European Medical Device Regulation (MDR)
MDR (Regulation (EU) 2017/745 on medical devices) is a new set of regulations governing medical device manufacturing and distribution within the European market. The new directive is a comprehensive overhaul of the MDD of 1993 and responds to the growing need for greater transparency of medical device compliance and fills gaps in an obsolete set of requirements.
As a regulation, MDR harmonizes standards on medical device compliance across all member states and is mandatory for all companies that wish to sell or distribute their products in the European marketplace.
Scope Of The Regulation
A medical device is defined as a product used to:
- Diagnose, prevent, monitor, treat or alleviate disability or injury
- Investigate, replace, or modify anatomical, physiological, and pathological processes
- Provide data for in-vitro examination of human samples
MDR puts significant importance on a lifecycle approach to medical device regulation and deviates slightly from the traditional focus on the pre-approval stage. From a compliance perspective, it means medical device manufacturing companies’ efforts must be deployed throughout the life of their products.
Of the most important changes from MDD to MDR, Annex XVI is arguably the section that heavily affects organizations in scope. Products that were not previously considered medical devices now fall under scrutiny, such as cosmetic products or products not intrinsically medical in nature.
There are also important changes in terms of class, where devices have been upgraded to a higher risk category and hereby must meet more stringent requirements.
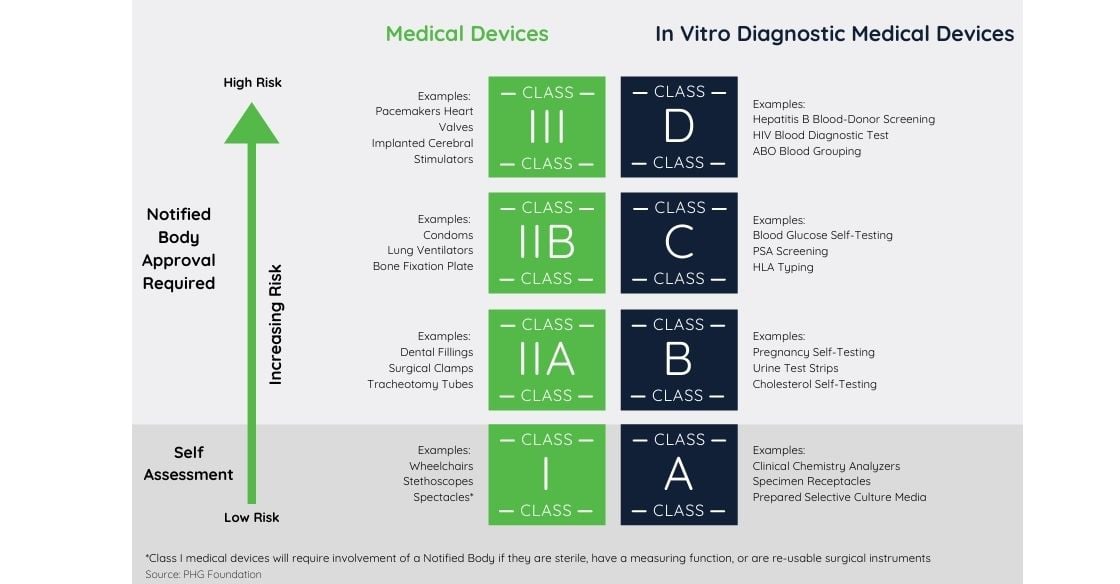
As already detailed in the directive 93/42/EEC on Medical Devices (MDD), risk classes are as follows:
- Class I-Devices: low risk such as non-invasive devices (stethoscopes, bandages, etc.).
- Class IIa-Devices: low-medium risk devices that are in the body between 60 minutes and 30 days.
- Class IIb-Devices: medium-high risk paused by devices in the body for more than 30 days (such as intensive care equipment).
Class III These are high-risk devices like prosthetic heart valves These go through a more comprehensive examination and testing process by the European Notified Body.
Traceability And Transparency
For all medical devices covered by MDR, the EU emphasizes the importance of traceability and introduced a mandate for Unique Device Identification (UDI), along with a Production Identifier (PI) for each batch of production.
Clinical investigations and pharmacovigilance are two other important aspects of MDR. Data on medical devices is made available to stakeholders, notified bodies, regulators, and consumers via the EUDAMED database.
From Directives To Regulation: Active Implantable Devices And In Vitro Diagnostic Medical Devices
The 1990 Council Directive 90/385/EEC on Active Implantable Medical Devices (AIMDD) defines in-scope products as devices that depend on exterior energy sources to operate and are completely introduced in the human body or placed at the surface of the eye by surgery. They are meant to stay in place in the body.
These devices and their accessories (batteries, programs, software) belong to the highest risk category and are subject to rigorous controls pre and post-market. Clinical experience and clinical use generate data that is exploited to determine the performance of the device:
- Published data with the device or equivalent
- Clinical investigation (or with a similar device)
- In vitro diagnostic medical devices
Directive 98/79/EC (IVDD) has been legally moved under the umbrella of a European regulation that will be applicable on May 2022 known as IVDR.
In vitro diagnostic medical devices are devices used to perform tests on human samples and designed to detect and diagnose a medical condition or monitor the efficacy of therapies.
The directive addresses the risks associated with the device based on sensitivity, diagnostic specificity, accuracy, repeatability, and reproducibility.
Medical Device Manufacturing Compliance In 2022 And Beyond
Regardless of the type of device, most directives are being phased out and becoming part of regulations better responding to the objective of increasing safety and protecting health.
Manufacturers must implement and document a quality system that shall remain effective throughout the lifecycle of the device, including procedures with document storage, post-market vigilance reports, and risk assessment.
Manufacturers are required to include a quality manual and all written procedures and policies regarding:
- Quality objectives
- Business organization
- Control, verification, validation, and review procedures during the design stage
- Quality assurance procedures during the manufacturing stage
- Relevant tests and trials
Simplifying Medical Device Compliance In The European Market
May 26, 2024, will be the last date to place non-compliant medical devices and in vitro devices on the European market with a deadline of May 26, 2025, to put them into service.
For AIMD, the new MDR requirements per class of products is changing the game to gain EU market access.
As the demand for smart and safe devices grows to offer better healthcare through innovative solutions, so does the competition. The one-year grace period the EU granted due to the pandemic should not have been a reason to become complacent. Making the required changes and gathering the data needed on your products, components, and substances will be time-consuming to say the least.
Navigating the compliance requirements on medical devices can be very complex and opaque supply chains can make the task nearly impossible.
Source Intelligence makes this process automated by connecting you to the supply chain data you need through our cloud-based platform. Whether you’re just entering the market or are preparing for the next regulatory deadline, our EU MDR and IVDR compliance solution offers you the best features around:
- AI-powered document verification so you only work with accurate data
- Automated reporting with document compilation and centralized storage
- Identification of data gaps and risks to help you prioritize steps needed
- Exclusive access to regulatory experts (PhDs and scientists), so you’re always up to date with requirements and developments
Request a demo to see what our EU MDR and IVDR solutions can do for you.
