


In May 2017, the EU launched two new medical technology regulations after an extensive overhaul of the directives governing the markets of Medical Devices (MDR) and In Vitro Diagnostic Devices (IVDR), EU 2017/745 and EU 2017/746 respectively.
As of May 26, 2021, the EU MDR is legally binding. A date of May 26, 2022 has been set for the application of EU IVDR. Considering the expansive work to be done to prepare, the next few will serve as a transition period.
Broadly speaking, the goals of EU IVDR and MDR aim to increase the safety of medical devices, improve traceability, and greatly strengthen post-sale vigilance over the entire lifecycle of the product.
In this article, we review the general principles of the directives, the most important requirements for IVD and medtech manufacturers, and how to overcome the obstacles of gathering supplier documentation.
EU IVDR And MDR Briefly
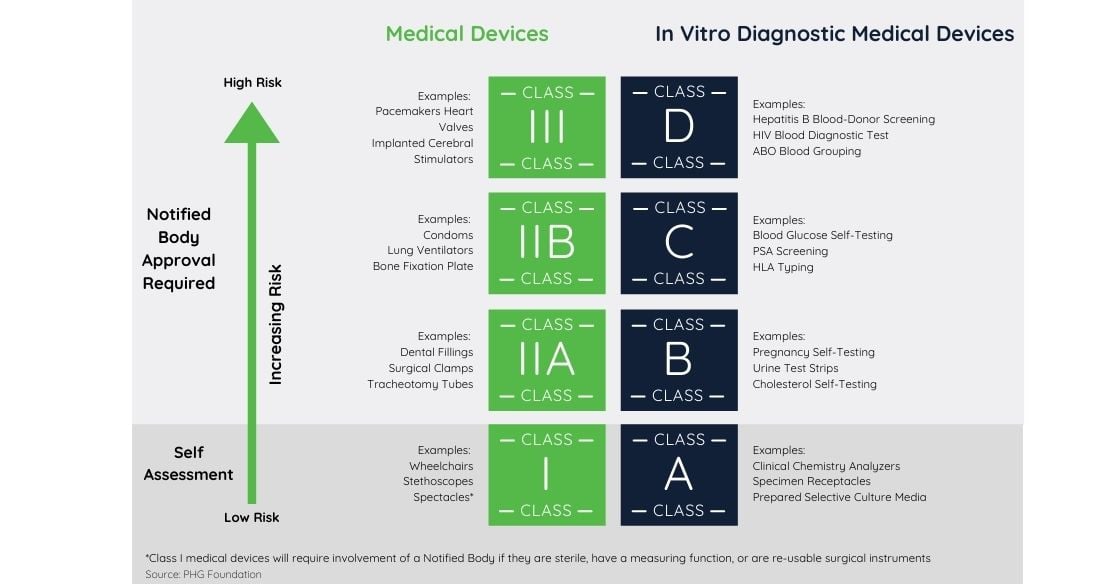
Medical devices are categorized by the risk they pose to human health, Class III representing the highest risk. Per the new EU medical device regulation, some non-medical devices have been added to the scope of the regulation including cosmetic fillers, products used to clean and sterilize, epilation lasers, and colored contact lenses. Other devices have been deemed to present higher risks than previously stated and now belong to a different class with stricter standards.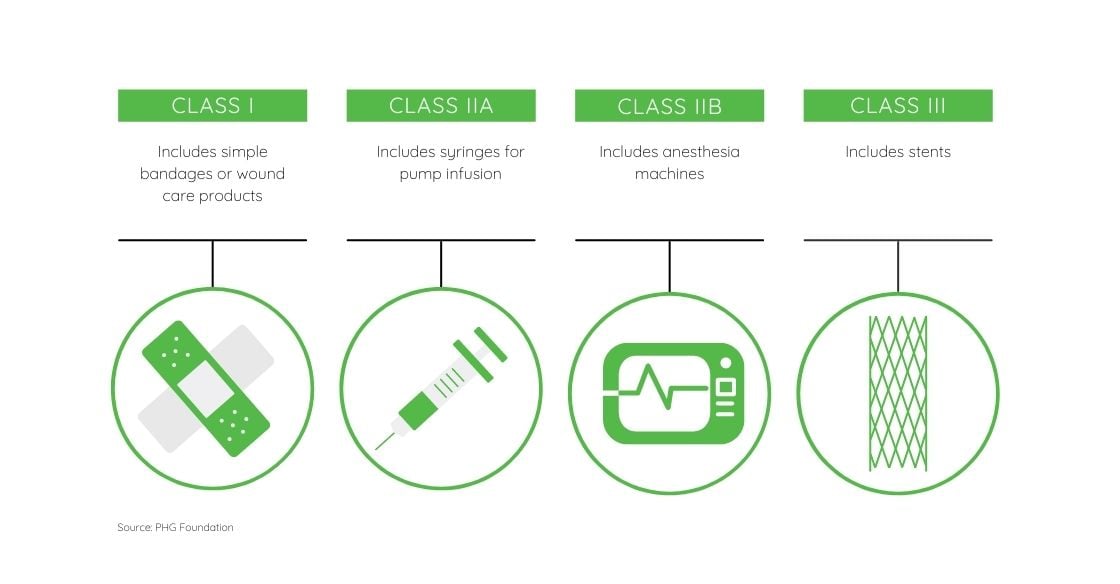 The EU Commission released a guide that breaks down the classifications and annexes to show where each medical device would fall into scope.
The EU Commission released a guide that breaks down the classifications and annexes to show where each medical device would fall into scope.

In-vitro diagnostic devices belong to either one of four categories in which the risk level goes from A (minor) to D (severe).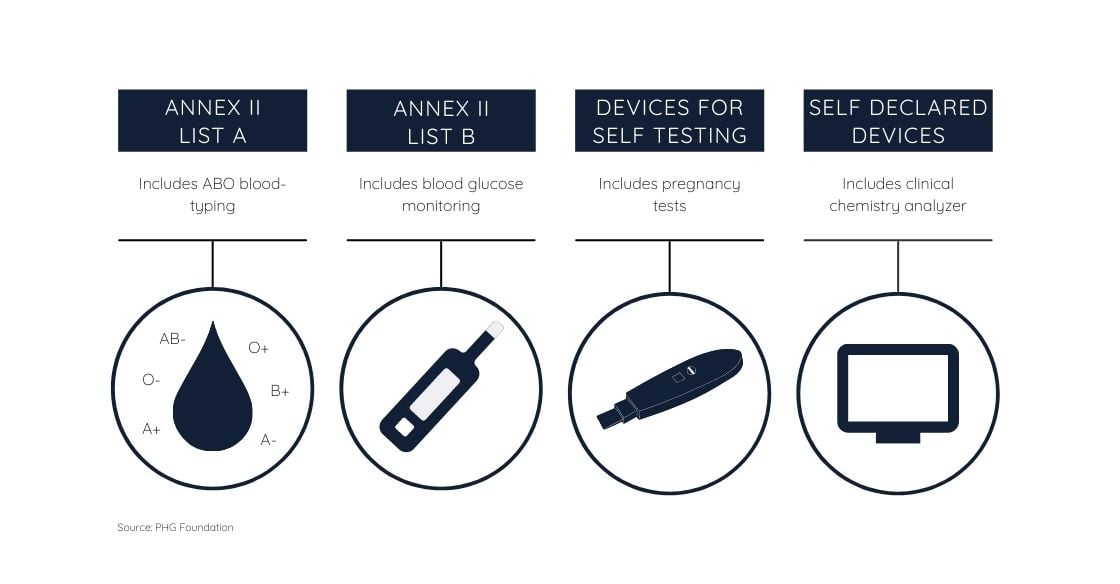 For devices presenting moderate to high risk, approval by a notified body is required.
For devices presenting moderate to high risk, approval by a notified body is required.
The regulations require duty-holders to implement or improve risk management and quality management systems. Conducting a clinical evaluation for each device is expected, along with providing all relevant technical documentation.
Manufacturers are also tasked with registering products with the EUDAMED database comprising of different modules related to:
- actor registration,
- unique device identification (UDI) and device registration,
- notified bodies and certificates,
- clinical investigations and performance studies,
- vigilance and market surveillance
Companies have the responsibility to upload and maintain/update all the data pertaining to chemical, physical and biological properties and disclose the presence of SVHCs in the device.
To increase traceability and transparency, devices will each wear a unique identifier (UDI) and be granted CE marking following the declaration of conformity.
Post-market surveillance is also an item of intense focus, which will likely prompt companies to review their incident reporting systems and processes.
Organizations must designate a person responsible for compliance and ensure full access to data and documentation.
Post-market vigilance (the process that confirms benefits and risks are acceptable by systematically reviewing and validating data) will be conducted in joint efforts from various stakeholders:
- The manufacturers, importers, and distributors will operate through data submission and reporting systems
- Member States will conduct surveillance via the EUDAMED database
- The European Commission will use the CIRCABC portal (Communication and Information Resource Center for Administrations, Businesses and Citizens), a web-based application.
EU IVDR And EU MDR Provisions For Specific Device Types And Activities
Among the 100 plus articles of the medical device directives (each), there are provisions concerning types of devices and regulatory activities. We’ll only mention a few and not dig into the specifics, but bear in mind the complexity and stringent requirements of the law.
Some of the provisions for devices include:
- Custom-made devices such as prosthesis or orthopedic footwear
- Investigational devices (subjects of clinical studies)
- Devices containing animal or human substances
- System and procedure packs
- Components and parts
Related Activities:
- UDI requirements
- Compliance with common specifications and harmonized standards
- New ethical requirements
EU IVDR And MDR Key Dates
May 26, 2021: EU MDR application
May 26, 2022: EU IVDR application
May 26, 2022: EUDAMED database to go live
May 26, 2024: MDD and IVDD certificates become void. Last date to place non-compliant products on the European market
May 26, 2025: last date MDD and IVDD products can be put into service
To help navigate the complex path to compliance, organizations can consult the guidance document established by the MDGC (Medical Device Coordination Group Document).
EU IVDR And MDR Compliance Solution
Beyond the fact that EU MDR has officially kicked off for Class III and implantable devices and the fast-approaching dates to fully roll-up data and meet requirements, the European regulations are a matter of long-term compliance.
The industry is a dynamic hub of advances and progress. Many products have only a 2-year lifecycle due to becoming outdated by better-performing ones. In 2019, the European Patent Office (EPO) received more than 13,800 patent applications in medical technology only. [4]
To keep up with the pace and not risk falling behind, companies have to implement strong mechanisms that facilitate the collection of data: recording, processing, validating, storing, and reporting functionalities are necessary to meet regulatory duties accurately and timely.
Source Intelligence offers an automated and user-friendly solution to simplify EU IVDR and MDR data collection and reporting.
With our cloud-based platform, your suppliers can easily upload data and documentation for the products that fall under the regulation’s scope. Our centralized solution makes it easy to identify gaps and access files as needed. To further ensure data accuracy, our proprietary AI technology aggregates the data it receives to deliver only validated information.
In Europe, more than half a million medical technologies are available, as reported by MedTech Europe in 2019. This figure represents a colossal number of suppliers who will get busier and busier as requests pour in.
Give your business a chance to improve responsiveness and engagement through a solution that benefits you and adds value to your supply chain. Request a demo of Source Intelligence’s EU IVDR and MDR program. Our compliance experts will show you how much you will gain from an affordable, exhaustive, and automated software solution.
